
A user-friendly technology
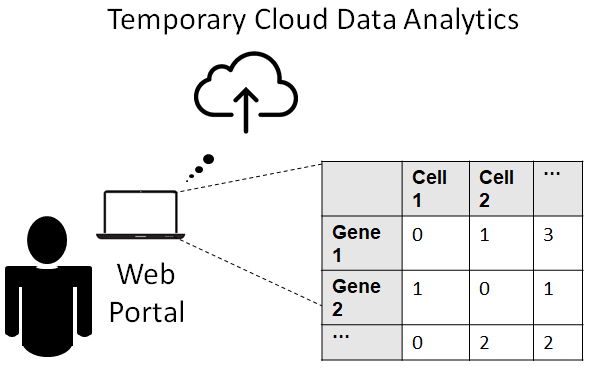 |
Uploading Data is as Easy as 1-2-3:
Researchers can directly upload their data of UMI counts to the web portal as a (gene x cell) matrix stored in a .csv, .txt or .rds file. Our cloud service does not store the user’s data or use it for any other purpose besides denoising. |
|
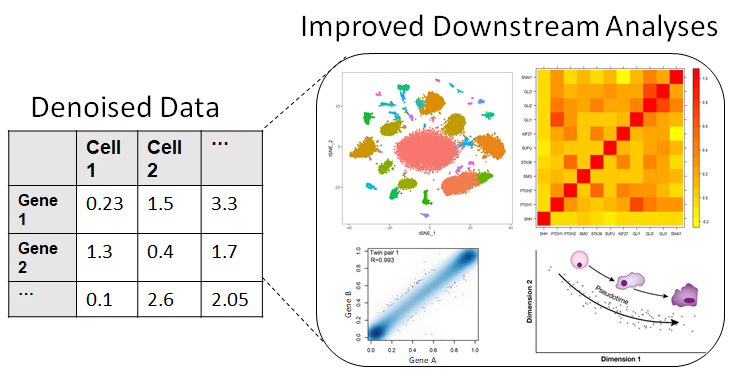
Use the denoised data for enhanced biological discovery:
After a short wait, the user will receive an email on the address they used to create an account. They can follow the link in the email to download the denoised data back. The link remains active for 96 hours. |
We encourage you to read our pre-print manuscript for more information!
Test Datasets
We provide demo datasets that users can avail of in order to test our web portal:
- Down-sampled data (1000 cells) from La Manno et al. 2016: Human Midbrain,
- Nine hundred purified immune cells from 10x Genomics: Human PBMCs, and
- Down-sampled mouse retina data (2000 cells) from Shekhar et al. 2014: Mouse Retina.
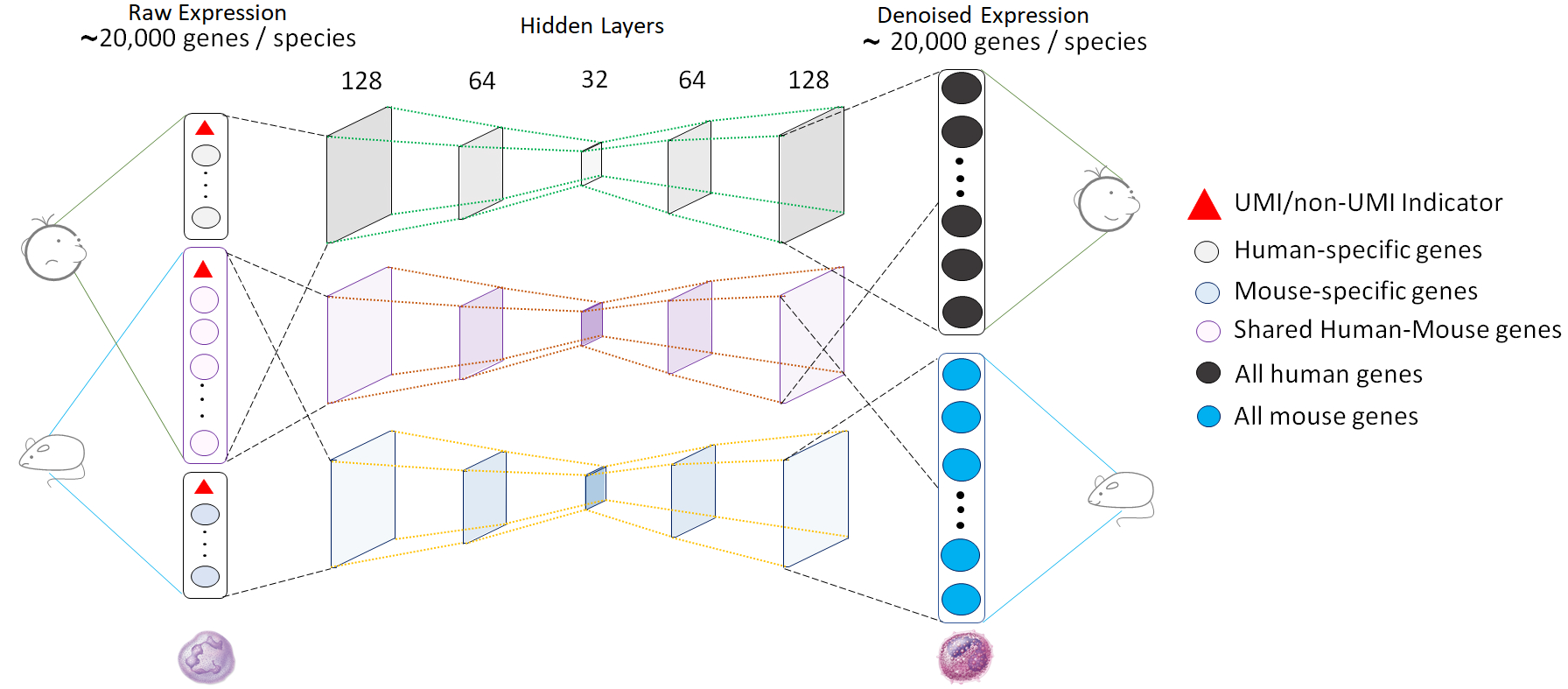
In most single cell transcriptomics (scRNA-seq) studies, the data are extremely sparse and noisy, hindering downstream analyses. To address this problem, we have developed Single-cell Analysis Via Expression Recovery via harnessing eXternal data (SAVER-X), a method for gene expression denoising and imputation. We leverage publicly available datasets and employ transfer learning for high quality scRNA-seq data denoising across a variety of settings. The network architecture for the auto-encoder used in our model is shown below. After implementing the auto-encoder, we perform a gene filtering step followed by Bayesian shrinkage.
Step 1: Register for an account
If you haven't already created an account, you should be seeing a page like this one:
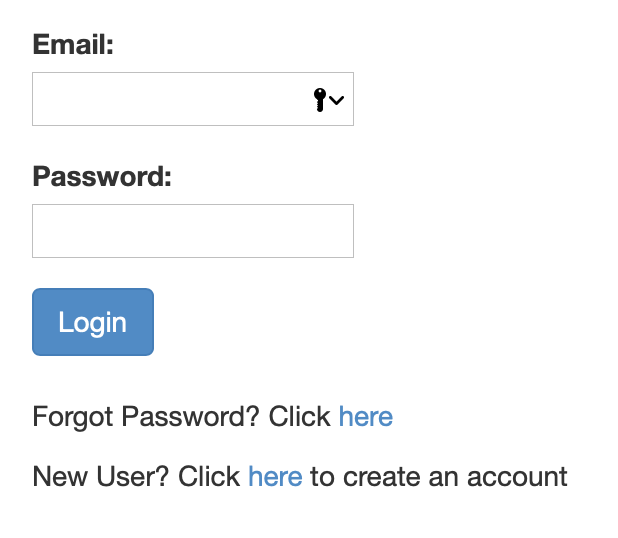
Simply follow the link and register for an account. You will be receiving notifications about the completion of your job, as well as the link to access the denoised data at the email address you use to create your account.
Step 2: Log in to your account
Once you successfully login, the screen should look like this:
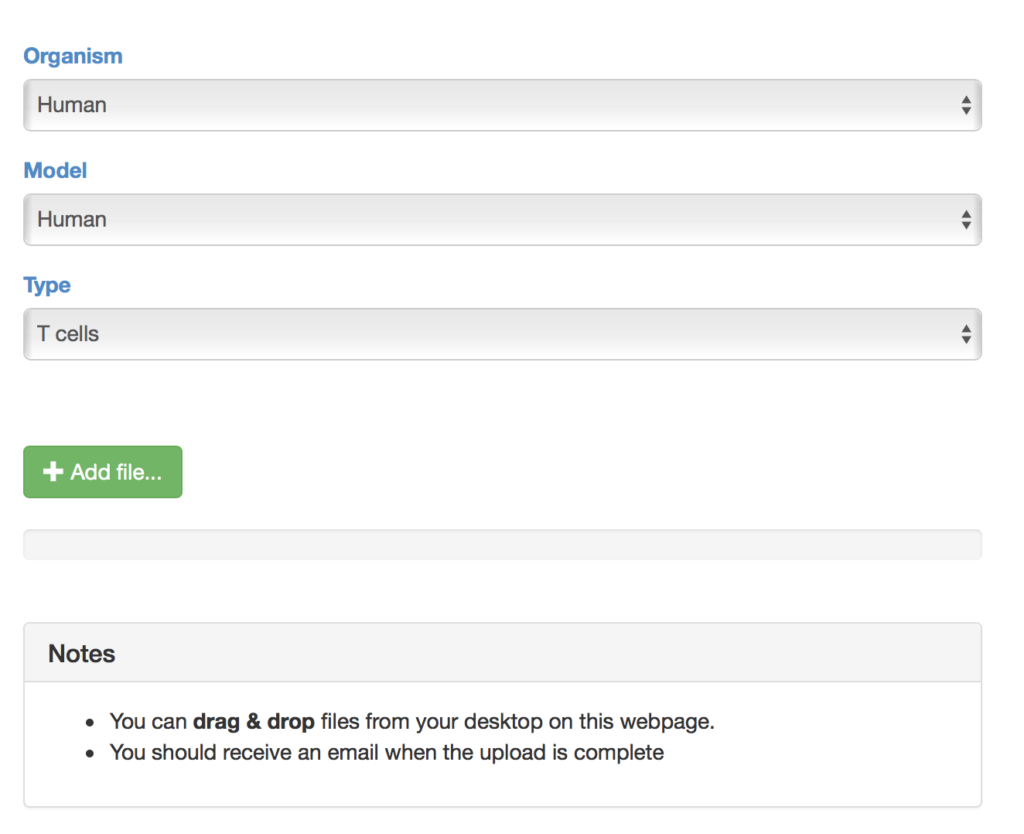
Step 3: Explore the SAVER-X settings that might help denoise your data
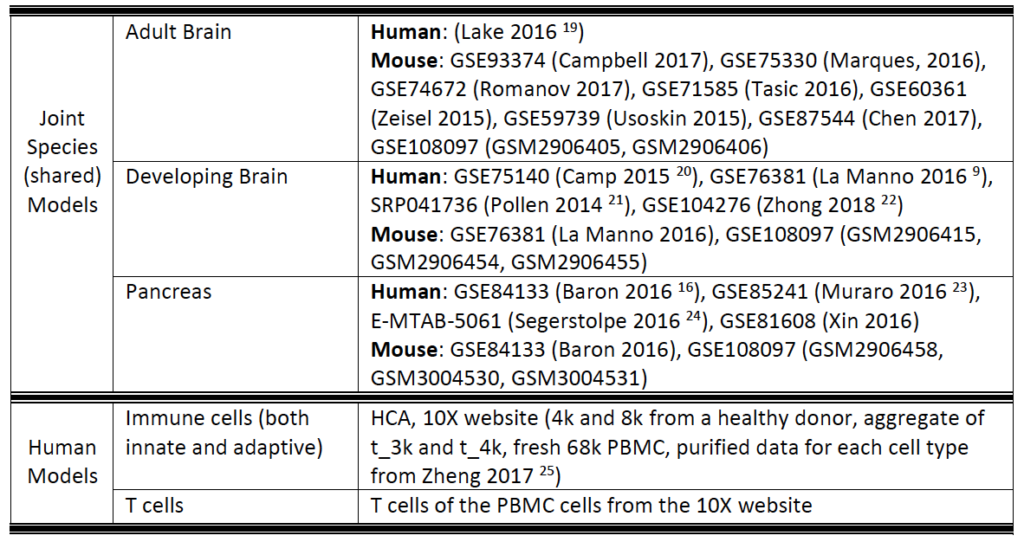
Currently, SAVER-X has trained deep count auto-encoder models across a range of tissue types (retina, brain, blood, etc.), cell types, as well as species. SAVER-X is equipped to deal with UMI counts, and the input matrix MUST BE a matrix of UMI counts. Users can choose a specific model involving any combination of these three variables against which they would like to denoise their data. Many of these choices will be context-dependent though they can also certainly be exploratory. For instance, if a researcher has generated single cell transcriptomics data from the mouse hippocampus, selecting "Mouse" and "Brain" would be the logical first choices. On the other hand, if you have generated data from the human retina but are interested to see if denoising it using a model trained on mouse retina can lead to new discoveries, then SAVER-X allows the users to explore such transfer learning questions. If you are unsure of how to select the models, "No Pretraining" is a default model choice.
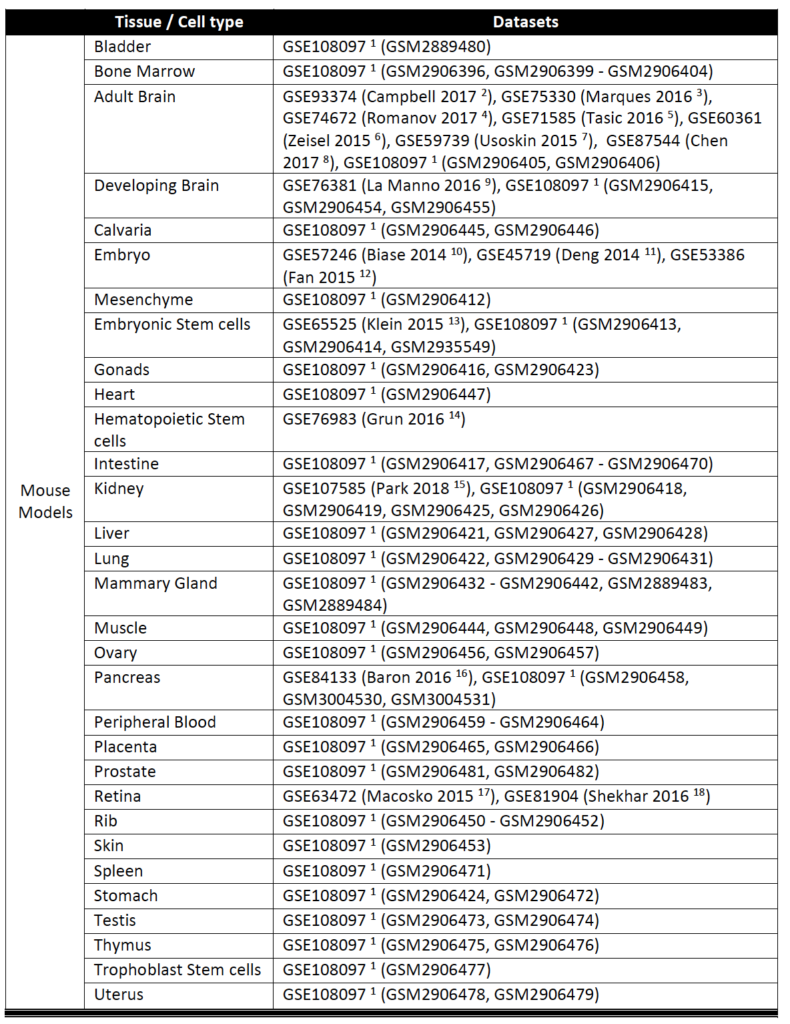
1. How large of a file can I upload?
The formal upload limit is 8GB, and our computation RAM limit is 32GB. You will get an out-of-memory error if you try to denoise a single file that violates the maximum permissible limit (32 GCB RAM). A typical batch with less than 8,000 cells would not exceed our current RAM limit. Matrices with more cells are also allowed on SAVER-X if the "No Pretraining" option is selected.
2. Why do I have to create an account? Is there limit to how much a single account can use the portal?
Creating an account is one of the ways our portal becomes more secure, and also allows to gauge how many users we accrue in the coming months. You can denoise as many files, and use the portal as frequently as you’d like. The limit will only be your curiosity, and funding $$$ of course.
3. How long will it take to get back denoised results?
Depends on how large the file size is. Typically, denoising a dataset of 1,000 cells takes less than 4 hours, and a dataset of 10,000 cells takes under 24 hours. You will receive an email with the link to download your denoised data as soon as the denoising has finished.
4. If I’m uploading my data to a web-tool, am I by default providing you consent to use my data?
No! We do not access your data in any way or use it for any internal purpose. We are using Amazon’s Big Data Analytics capabilities for our portal, and the S3 Buckets used to store the data objects get “emptied” every few days.
5. What is the recommended file format, again?
Your data must be uploaded as a gene x cell matrix, stored in a .csv, .txt or .rds file. For the .rds file format, the data can either be stored as a regular matrix or sparse matrix (provided by the R package: Matrix).
6. Why do you have both Immune cells and T cells under the Human model?
SAVER-X trained on “Immune cells” includes a general model trained on virtually all subtypes of innate and adaptive immune cells. The “T cells” model is more specific, so if your study has performed FACS sorting, or is specifically looking at only T cells, then the latter model might be more appropriate. Otherwise, a general model would also be able to robustly denoise your dataset.
7. What if I have performed scRNA-seq on an organism (who doesn’t want to sequence the shark rectal gland?) that you don’t currently have a pre-trained model for?
Well, you’re still in luck. While we may not have every single species and organ listed at the moment, you can do one of two things. First, use the “No Pretraining” model. This would still give you improved, denoised values; it just won’t have much existing data to harness from while denoising your data. Second, explore related organs/species. To do so, map the genes that are shared between sharks and mice or humans. You can denoise homologous genes using either an existing mouse or human pretrained SAVER-X model. We imagine that you likely sequenced the rectal gland to study osmoregulation, or how tubules work to transport ions and water. If so, you could certainly experiment by using a model pre-trained on the human kidney or the intestine, and explore how informative the results are. After all, isn’t this the true power of transfer learning?
8. Are some pre-trained models better at denoising than others? If so, why?
We tried to bring out the best in every model, but some pre-trained models start out with an advantage if several large studies have already sequenced that organism/tissue of interest. Models that are pre-trained on enormous, high-quality datasets tend to slightly edge out those models trained on fewer cells. For instance, our Human Immune Cells model is pre-trained on ~1 million cells, a combination of datasets obtained from the Human Cell Atlas and 10x Genomics, whereas the Mouse Rib model is trained on less than 25,000 cells. Therefore, the former pre-trained model is more efficient than the latter. We do strive, however, to update each model as newer publicly obtainable datasets become accessible.
9. I have more questions and/or want to provide feedback, who do I contact?
Please email singlecell@wharton.upenn.edu. We strive to resolve all queries within 72 hours. We would also love to hear your feedback on how your experience was in using our web portal.
Ready to denoise?
Below is a form in which you can upload a .csv, .txt or .rds file containing your data, and run it against the SAVER-X model most appropriate for your data.